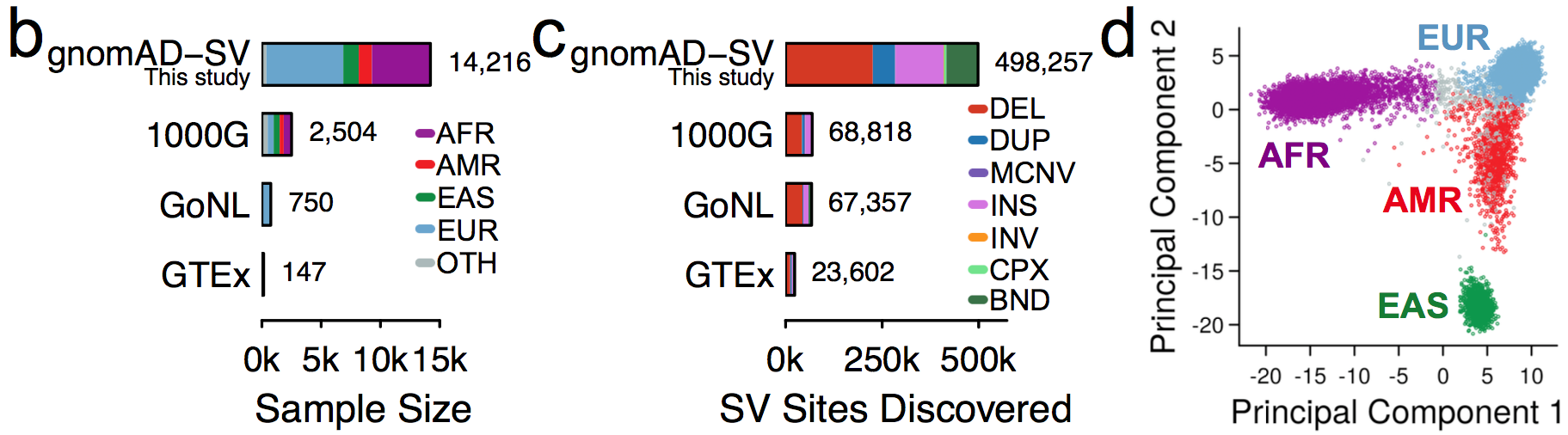
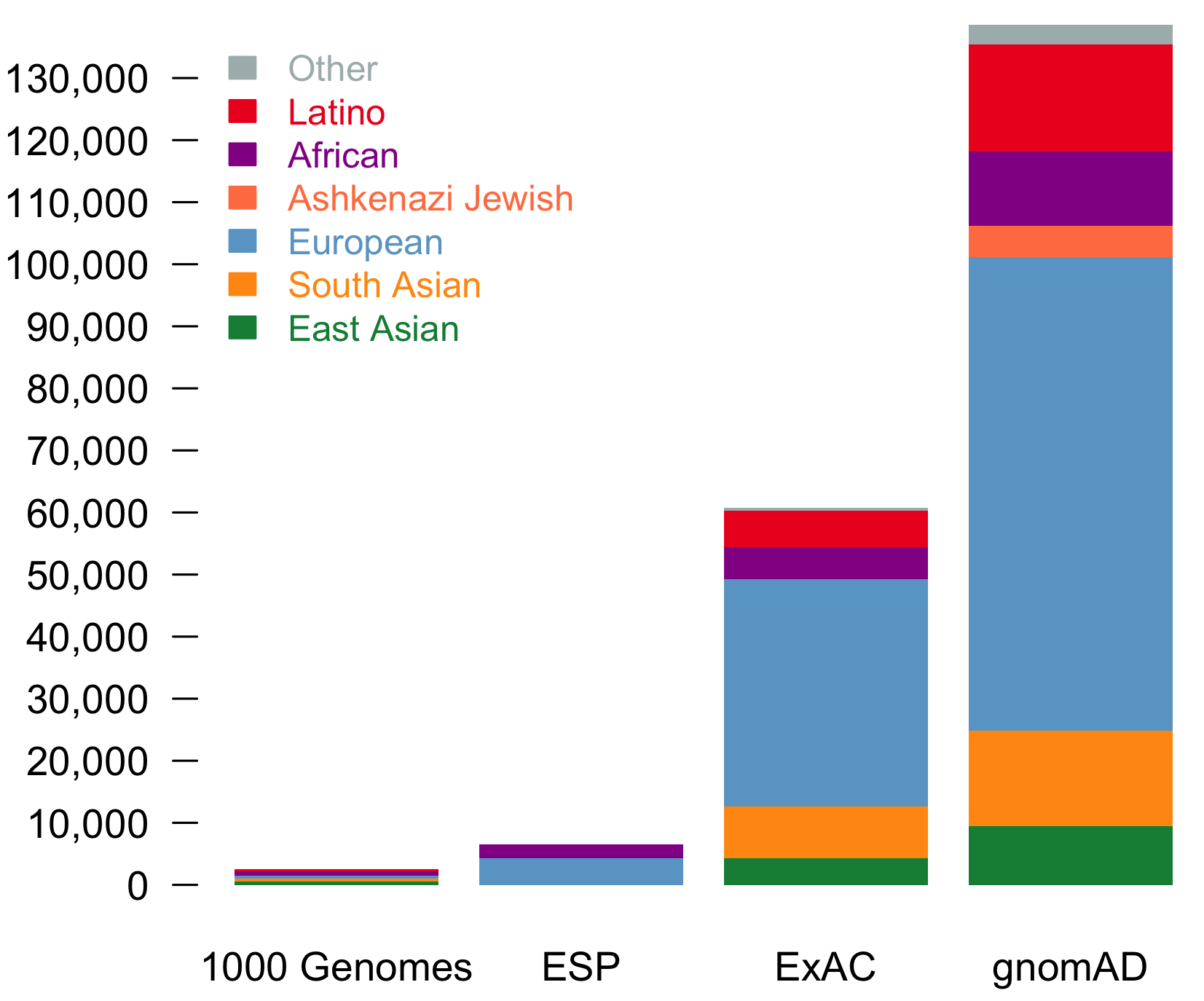
It is an absolute pleasure to announce the official release of the gnomAD manuscript package. In a set of seven papers, published in Nature, Nature Medicine, and Nature Communications, we describe a wide variety of different approaches to exploring and understanding the patterns of genetic variation revealed by exome and genome sequences from 141,456 humans.
Publication announcements always feel a little strange in this new era of open science. In our case, the underlying gnomAD data set has been publicly fully available for browsing and downloading since October 2016, and we’ve had the preprints available online since early 2019. However, it’s undeniable that there is something deeply gratifying about seeing these pieces of science revealed in their final, concrete form.
For me this package has a particular significance – it represents the culmination of seven and a half years of work with a phenomenal team at the Broad Institute, and marks my transition to a new role in Australia, and the handover of the gnomAD project to new leadership. So I wanted to spend some time in this post reflecting on the history of the project that became gnomAD, the people who’ve made it possible, and where things will go from here.
Continue reading The gnomAD papers