Laurent Francioli & Daniel MacArthur
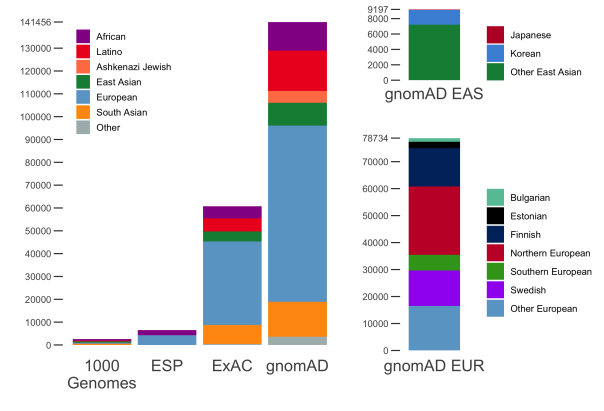
We are thrilled to announce the release of gnomAD v3, a catalog containing 602M SNVs and 105M indels based on the whole-genome sequencing of 71,702 samples mapped to the GRCh38 build of the human reference genome. By increasing the number of whole genomes almost 5-fold from gnomAD v2.1, this release represents a massive leap in analysis power for anyone interested in non-coding regions of the genome or in coding regions poorly captured by exome sequencing.
In addition, gnomAD v3 adds new diversity – for instance, by almost doubling the number of African-American samples we had in gnomAD v2 (exomes and genomes combined), and also including our first set of allele frequencies for the Amish population.