As we were organizing analyses for the ExAC flagship paper, we were inspired by Titus Brown’s manifesto on reproducibility. As with most collaborative papers, we had a core team of analysts, each responsible for their subset of analyses and figure panels. Part of the way through, we realized there were many shared features that we wanted to keep consistent across analyses, particularly variant filtering strategies and annotations. We made the decision to organize the code in a central github repository, which the team members could access and edit as analyses progressed. Today, we are happy to release this code, which you can use to reproduce every figure in the paper!

How to use
The code and miscellaneous data are available at https://github.com/macarthur-lab/exac_2015.
git clone git@github.com:macarthur-lab/exac_2015.git
cd exac_2015Then, start R, and run the following. Warning: this process requires a decent amount of memory, ideally on a machine that has at least 16G RAM (newer Macbook Pros are fine, but not Macbook Airs). On Linux and machines with stricter memory policies, we recommend 32G or even 64G to allow for overhead in loading.
source('exac_constants.R')
exac = load_exac_data()## [1] "Using locally loaded exac file. Run exac = load_exac_data(reload=T) to update to newest file."
## [1] "Step (1/6): Loading data..."
## [1] "Now processing data (2/6): Determining sequence contexts..."
## [1] "(3/6): Computing allele frequencies..."
## [1] "(4/6): Calculating bad regions of the genome..."
## [1] "(5/6): Determining what to _use_..."
## [1] "(6/6): Parsing SIFT and PolyPhen scores, and separating functional categories..."
## [1] "Done! Here you go. Took:"
## user system elapsed
## 857.437 17.924 878.059This will take about 10-15 mins to load, plus a ~800M download the first time you run it. exac is then the data frame of ALL ~10M variants in the dataset with over ~100 annotations per variant. Each variant is now its own entry, as opposed to a typical VCF where multi-allelic variants are combined into a single line. Note that these are unfiltered variant calls. Make sure to filter to either PASS sites, or better yet, our criteria for high-quality calls (given by the column use, see below for details):
filtered_calls = subset(exac, filter == 'PASS')or:
use_data = subset(exac, use)You can view the table using head() or with dplyr’s tbl_df, which formats the output nicely:
tbl_df(exac)## Source: local data frame [10,195,872 x 116]
##
## chrom pos ref alt id filter
## (chr) (int) (chr) (chr) (chr) (chr)
## 1 1 13372 G C . PASS
## 2 1 13380 C G . VQSRTrancheSNP99.60to99.80
## 3 1 13382 C G . VQSRTrancheSNP99.60to99.80
## 4 1 13402 G C . VQSRTrancheSNP99.60to99.80
## 5 1 13404 G A . VQSRTrancheSNP99.60to99.80
## 6 1 13404 G T . VQSRTrancheSNP99.60to99.80
## 7 1 13417 C CGAGA . PASS
## 8 1 13418 G A . AC_Adj0_Filter
## 9 1 13426 A G . VQSRTrancheSNP99.80to99.90
## 10 1 13438 C A . VQSRTrancheSNP99.80to99.90
## .. ... ... ... ... ... ...
## Variables not shown: ac (int), ac_afr (int), ac_amr (int),
## ac_adj (int), ac_eas (int), ac_fin (int), ac_hemi (int),
## ac_het (int), ac_hom (int), ac_nfe (int), ac_oth (int),
## ac_sas (int), ac_male (int), ac_female (int),
## ac_consanguineous (int), ac_popmax (int), an (int), an_afr
## (int), an_amr (int), an_adj (int), an_eas (int), an_fin
## (int), an_nfe (int), an_oth (int), an_sas (int), an_male
## (int), an_female (int), an_consanguineous (int), an_popmax
## (int), hom_afr (int), hom_amr (int), hom_eas (int), hom_fin
## (int), hom_nfe (int), hom_oth (int), hom_sas (int),
## hom_consanguineous (int), clinvar_measureset_id (int),
## clinvar_conflicted (int), clinvar_pathogenic (int),
## clinvar_mut (chr), popmax (chr), k1_run (chr), k2_run
## (chr), k3_run (chr), doubleton_dist (dbl), inbreedingcoeff
## (dbl), esp_af_popmax (dbl), esp_af_global (dbl), esp_ac
## (int), kg_af_popmax (dbl), kg_af_global (dbl), kg_ac (int),
## vqslod (dbl), gene (chr), feature (chr), symbol (chr),
## canonical (chr), ccds (chr), exon (chr), intron (chr),
## consequence (chr), hgvsc (chr), amino_acids (chr), sift
## (chr), polyphen (chr), lof_info (chr), lof_flags (chr),
## lof_filter (chr), lof (chr), context (chr), ancestral
## (chr), coverage_median (dbl), coverage_mean (dbl),
## coverage_10 (dbl), coverage_15 (dbl), coverage_20 (dbl),
## coverage_30 (dbl), pos_id (chr), indel (lgl),
## bases_inserted (int), transition (lgl), cpg (lgl),
## alt_is_ancestral (lgl), af_popmax (dbl), af_global (dbl),
## singleton (lgl), maf_global (dbl), daf_global (dbl),
## daf_popmax (dbl), pos_bin (fctr), bin_start (dbl), sector
## (chr), bad (lgl), use (lgl), lof_use (lgl), sift_word
## (chr), sift_score (dbl), polyphen_word (chr),
## polyphen_score (dbl), category (chr)
A previous blog post by Monkol Lek, A Guide to the Exome Aggregation Consortium (ExAC) Data Set, described some of the challenges in exploring the data. With the dataset loaded into R, we can dive into a few more tips and tricks. First, you’ll notice that there are many annotations per variant pre-calculated, in particular:
Allele frequency
In particular, one set of questions that always comes up is regarding the allele counts (AC) and allele numbers (AN). The raw counts (ac and an) refer to the total number of chromosomes with this allele, and total that were able to be called (whether reference or alternate), respectively. Thus, the allele frequency is ac/an. However, for more stringent applications, we adjusted the allele count to only include individuals with genotype quality (GQ) >= 20 and depth (DP) >= 10, which we term ac_adj (and correspondingly, an_adj for total number of chromosomes from individuals with those criteria regardless of genotype call).
In this table, af_global is then ac_adj/an_adj and ac and an from each population is filtered to only adjusted counts (i.e. ac_EAS includes only individuals of East Asian descent that meet this criteria). Additionally, we provide a maximum allele frequency across all populations as af_popmax and the corresponding population as popmax, which is useful for filtering variants for Mendelian gene discovery. Finally, we have hom_* for all populations, which is the count of homoyzgous individuals for this allele.
Functional annotations
The output from VEP is parsed for each allele and, in the default table, summarized using the most severe consequence across all transcripts. You can run load_exac_data('canonical') for instance to load the annotations for only the canonical transcripts, or pass transcript=T or gene=T to load in a data frame with one entry for every transcript or gene, respectively, for each variant.
Loss-of-function (LoF) annotations of variants from LOFTEE are provided in the lof, lof_filter, and lof_flags columns, and the highest-confidence variants are indicated as lof_use. PolyPhen and SIFT annotations are parsed into words (e.g. benign or possibly_damaging) and scores.
There are also annotations from ClinVar, intersected with output of Eric Minikel’s Clinvar parsing tool. A query for pathogenic variants without conflicting evidence may use clinvar_pathogenic == 1 && clinvar_conflicted == 0 && clinvar_mut == 'ALT'.
Other annotations
Finally, there are numerous convenience annotations for quick analysis, including logical values for indel (as well as the number of bases inserted or deleted in bases_inserted), cpg, transition, singleton, various coverage metrics (mean, median, and proportion covered at a given depth – e.g. coverage_30 indicates the proportion of individuals covered at 30X). The 3-base context around SNPs is provided in the context column (warning: this is currently broken for indels, and in any case, not as useful).
As mentioned above, the use column is a crucial one for filtering to the highest-quality variants, which includes the following criteria:
- Filter status of “PASS” (from VQSR)
- Adjusted allele count (ac_adj, see above) > 0 (i.e. there is at least one individual with at least GQ >= 20 and DP >= 10)
- At this locus, at least 80% of individuals (regardless of whether they have the variant) met the high-quality adjusted status as above (in code,
exac$an_adj > .8*max(exac$an_adj)) - Does not fall in the top 10 most-multiallelic kilobases of the genome (see Multi-allelic enriched regions)
With the data, there are many analyses that you can then do, including:
Recreate the figures from the paper
This one’s an easy one. Just run the following, which will take ~an hour. (Warning: will take lots of memory and CPU and has been known to crash R, particularly for some more computationally expensive figures, such as Figures 2E and 3F).
source('exac_figures.R')Depending on your system, it may be easier to open the script and run sections of the code independently.
Multi-allelic enriched regions
Early on after we released the ExAC dataset to the public, Nicole Deflaux at Google performed a nice analysis identifying chromosome 14 as a hotspot of multi-allelic variants (full analysis here: https://github.com/deflaux/codelabs/blob/exac/R/ExAC-Analysis/ExAC-Analysis.md). We wanted to trace the source of this issue, so we used ggbio to plot the multi-allelics across the genome.
Install ggbio if its not already installed:
source("https://bioconductor.org/biocLite.R")
biocLite("ggbio")Then, use the genome_plotting.R script where we’ve written functions to calculate and plot the density of multi-allelic variants (for now, limiting to quad-allelic and up) per kilobase across the genome.
library(ggbio)
source('analysis/supplement/genome_plotting.R')
multiallelic_density = get_multiallelic_density(exac, resolution=1000)
suppressMessages(plot_multiallelic_density(multiallelic_density))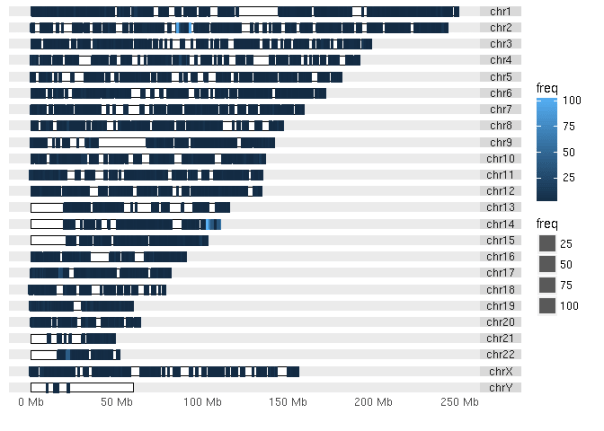
The 2 regions highlighted in light blue look interesting (chr14 as before, as well as chr2), so we can dig into those regions. The top 10 regions ordered by frequency are:
multiallelic_density %>%
arrange(desc(freq)) %>%
head(10)## chrom bin_start bin_end freq
## 1 14 106330000 106331000 109
## 2 2 89160000 89161000 103
## 3 14 106329000 106330000 76
## 4 14 107178000 107179000 63
## 5 17 18967000 18968000 46
## 6 22 23223000 23224000 44
## 7 1 152975000 152976000 42
## 8 2 89161000 89162000 39
## 9 14 107179000 107180000 38
## 10 17 19091000 19092000 38If we look these up, we’ll find that these regions include immunoglobulin kappa (IGK, chr2), heavy (IGH, chr14), lambda variable (IGLV, chr22), and joining (IGKJ1, chr2), as well as some difficult-to-call regions of the genome. In ExAC, we’ve added a column for whether a variant falls into these 10 regions of the genome, named bad for lack of a better phrase, and these variants are then excluded from the high-quality use variants.
dbSNP rs number versus allele frequency
We can also do some fun analyses that didn’t make the cut. One easy hypothesis we had was whether rs number (the numbers in a dbSNP RSID identifier) would correlate with allele frequency. We would expect a negative correlation, as rs numbers are sequentially assigned, and common variants should have been discovered earlier on. Of course, this will not be a perfect correlation, as rare variants in the first individuals sequenced, as well as disease variants, will be identified earlier and have lower rs numbers.
First, let’s get the rsid column into a numeric form and subset to only those in dbSNP:
library(magrittr)
library(tidyr)
exac %<>% mutate(rsid = as.numeric(gsub('rs', '', id)))
rsid_variants = filter(exac, !is.na(rsid) & af_global > 0)The rs number is negatively correlated with allele frequency, as expected.
cor.test(rsid_variants$rsid, rsid_variants$af_global)##
## Pearson's product-moment correlation
##
## data: rsid_variants$rsid and rsid_variants$af_global
## t = -536.42, df = 1108700, p-value = 0
## alternative hypothesis: true correlation is not equal to 0
## 95 percent confidence interval:
## -0.4554037 -0.4524481
## sample estimates:
## cor
## -0.4539271library(ggplot2)
ggplot(rsid_variants) + aes(x = rsid, y = af_global) + geom_point() + geom_smooth(method="lm", se=F) + ylab('Global Allele Frequency')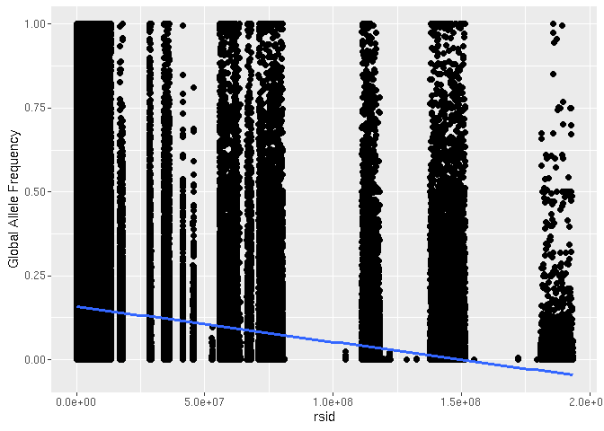
It’s a bit tough to see the correlation there, but it’s promising: later RSIDs do appear to have lower allele frequencies than earlier ones. Our fit line doesn’t seem too great, so let’s use the log of the allele frequency and use smoothScatter() to visualize it:
smoothScatter(rsid_variants$rsid, log10(rsid_variants$af_global), xlab='RSID', ylab='Global allele frequency (log10)', col='white')
abline(lm(log10(rsid_variants$af_global) ~ rsid_variants$rsid))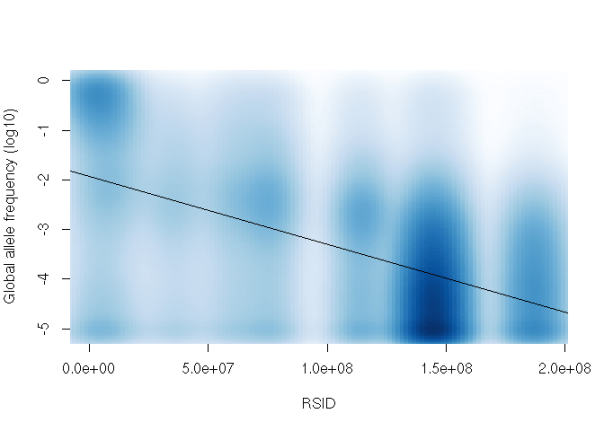
The largest blob in the bottom right corresponds to variants submitted by the NHLBI Exome Sequencing Project, from which many exomes are included in ExAC.
We encourage anyone to continue exploring the ExAC dataset and we hope that this repository helps others reproduce and expand on the analyses we’ve done. We welcome any and all comments, here and on the github page. Enjoy!

How do I generate a PCA plot from the ExAC data? Thanks.
LikeLike
buy Minocin no prescription needed
buy cheap generic Minocin
buy Minocin online in canada
non prescription cheap Minocin usa
buy discount Minocin online usa
cheap Minocin overnight
generic Minocin usa
discount pharmacy Minocin online
us pharmacy Minocin
cheapest Minocin in canada online order
sale Minocin online without prescription
LikeLike
Nice work making it easy to replicate your analysis. Is there any way get some kind of ID on the sample/source of a unique variant? I would like to know what other variants this sample contains…
Thank you
LikeLike
Amazing work guys. ExAc is Electric for Exome Analysis
LikeLike