Konrad Karczewski and Laurent Francioli
Today, we are pleased to announce the formal release of the genome aggregation database (gnomAD). This release comprises two callsets: exome sequence data from 123,136 individuals and whole genome sequencing from 15,496 individuals. Importantly, in addition to an increased number of individuals of each of the populations in ExAC, we now additionally provide allele frequencies across over 5000 Ashkenazi Jewish (ASJ) individuals. The population breakdown is detailed in the table below.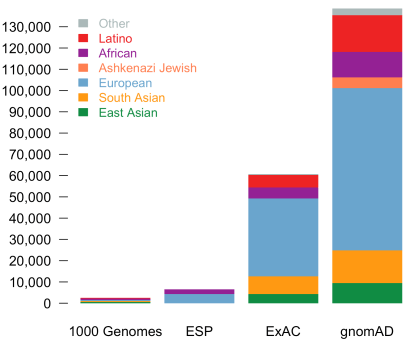
Continue reading The genome Aggregation Database (gnomAD)

