Ryan Collins, Harrison Brand, Daniel MacArthur, and Mike Talkowski
The first gnomAD structural variant (SV) callset is now available via the gnomAD website and integrated directly into the gnomAD Browser.
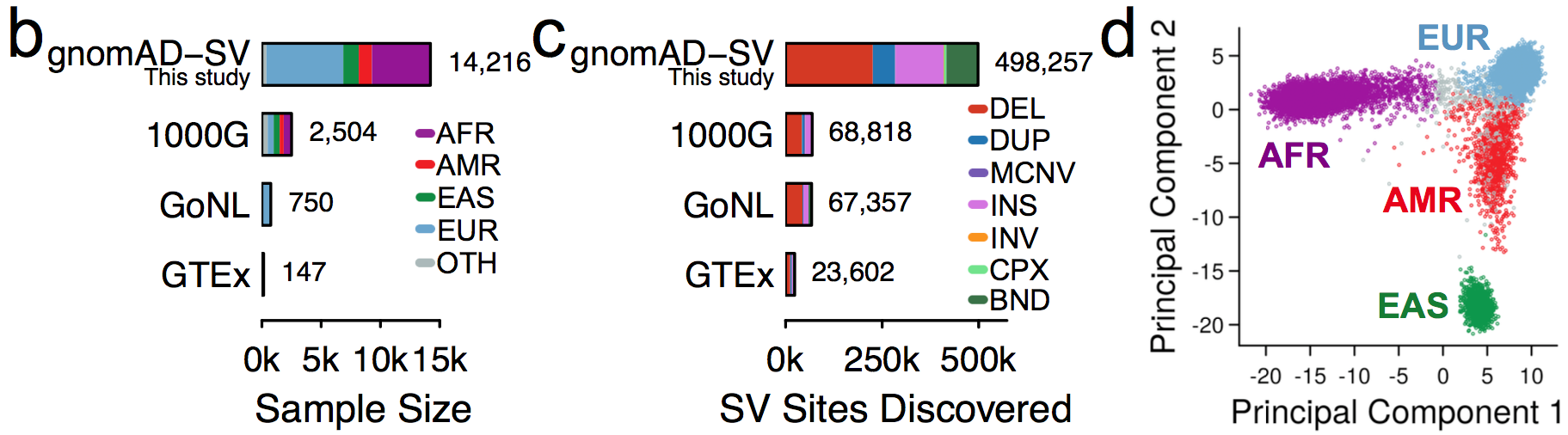
This initial gnomAD SV callset includes nearly a half-million distinct SVs across seven SV mutational classes and 13 subclasses of complex SVs detected in 14,891 genomes spanning four major global populations. In the publicly released callset and gnomAD browser, you can find site, frequency, and annotation data for ~445k SVs from 10,738 unrelated genomes with appropriate consent to allow the release of this information.
In this post we summarize how we created this new call set, and some important practical considerations when using it. You can get more details, including callset generation and analyses, in the full gnomAD-SV preprint available on bioRxiv.