We live in an amazing time to do human genetics. Over the last five years, thanks to impressive advances in DNA sequencing technology, the research community has collected sequencing data on genetic variation from over 200,000 samples. This provides us, for the first time, with the ability to study genetic variants at very low frequencies in the general population. However, in order to perform this research it’s critical that these genetic data be brought together and analyzed in the same way to ensure that the genetic changes that we find are real, and not the artifacts of differences in sequencing technology or analytical pipelines.
This goal is what drives the Exome Aggregation Consortium (ExAC), an international coalition of investigators with a focus on data from exome sequencing – an approach that allows us to focus variant discovery on the regions of the genome that encode proteins, known collectively as the exome. To date the Consortium has accumulated and jointly analyzed exome data from nearly 92,000 individuals, and has prepared a publicly accessible data set spanning 61,486 of these individuals for use as a global “reference set”. While the individuals in the reference set aren’t necessarily healthy – many have adult-onset diseases such as type 2 diabetes and schizophrenia – we have removed individuals with severe pediatric diseases, making this (we believe) a reasonable comparison data set for childhood-onset Mendelian diseases.
On October 20th at the American Society of Human Genetics (ASHG) conference we announced release 0.1 of the ExAC data set in two forms, as a browser and a downloadable raw data file. This was not just a massive data release but also a massive collaborative effort, which is detailed here. Four weeks after the release, the ExAC browser has received over 120,000 page views from over 17,000 unique users, and the raw data has been downloaded by over 150 organizations. The annotation tools ANNOVAR and ATAV have provided updates that have incorporated the ExAC data and the developers of Combined Annotation Dependent Depletion (CADD) have provided corresponding CADD scores. The commercial tools from GoldenHelix and GeneTalk have also incorporated the ExAC data. As the lead analyst on the project for over 2 years, I’ve been thrilled with the response it has received and the kind words and valuable feedback from the research community.
This practical guide, which uses two example genes FBN1 and MECP2 is aimed at general users and how they can access information using the ExAC Browser.
FBN1 Example
John Belmont commented on Nature News and Twitter that the FBN1 gene associated with Marfan syndrome has 11 subjects with Loss of function (LoF) mutations. If these are true disease causing variants then it fits roughly with the 1 in 5000 incidence of this disease.
The FBN1 LoF variants can be directly viewed on the ExAC browser by searching FBN1 or clicking on this link and then clicking on the LoF button. Things to note on the FBN1 gene summary page:
- The coverage plot (affectionately called the Guilin plots ) is of the canonical transcript, using the Ensembl definition. This may not necessarily correspond to the clinically relevant transcript.
- The genomic coordinates uses GRCh37 and NOT from the recently released GRCh38
- Variant sites with multiple alleles are represented on separate rows
- The functional annotation and corresponding protein consequence is from the most severe impact amongst the transcripts and may not affect all transcripts. As it is summarized from multiple transcripts, the amino acid position can sometimes appear out of order.
- Allele Number is the number of chromosomes so is twice the number of individuals (maximum 2*61,486). Due to the nature of exome capture and quality thresholds applied, this will not always be at the maximum.
- All variant data displayed in the table can be downloaded as a CSV text file and opened in Excel to restore the columns and rows.
Amongst the 10 Loss of Function variants in FBN1
Using the stop gained 15-48719948-G-C variant page is a good example to highlight important features:
- The histogram of Depth and Genotype Quality (GQ) for individuals with the allele or click on the full site metrics check box to display the histogram for all individuals with genotype calls (including those that are homozygous reference).
- The stop gain variant does not affect all transcripts. The variant results in a missense change in the canonical transcript (ENST00000316623). In fact the canonical transcript has only 8/10 LoF mutations.
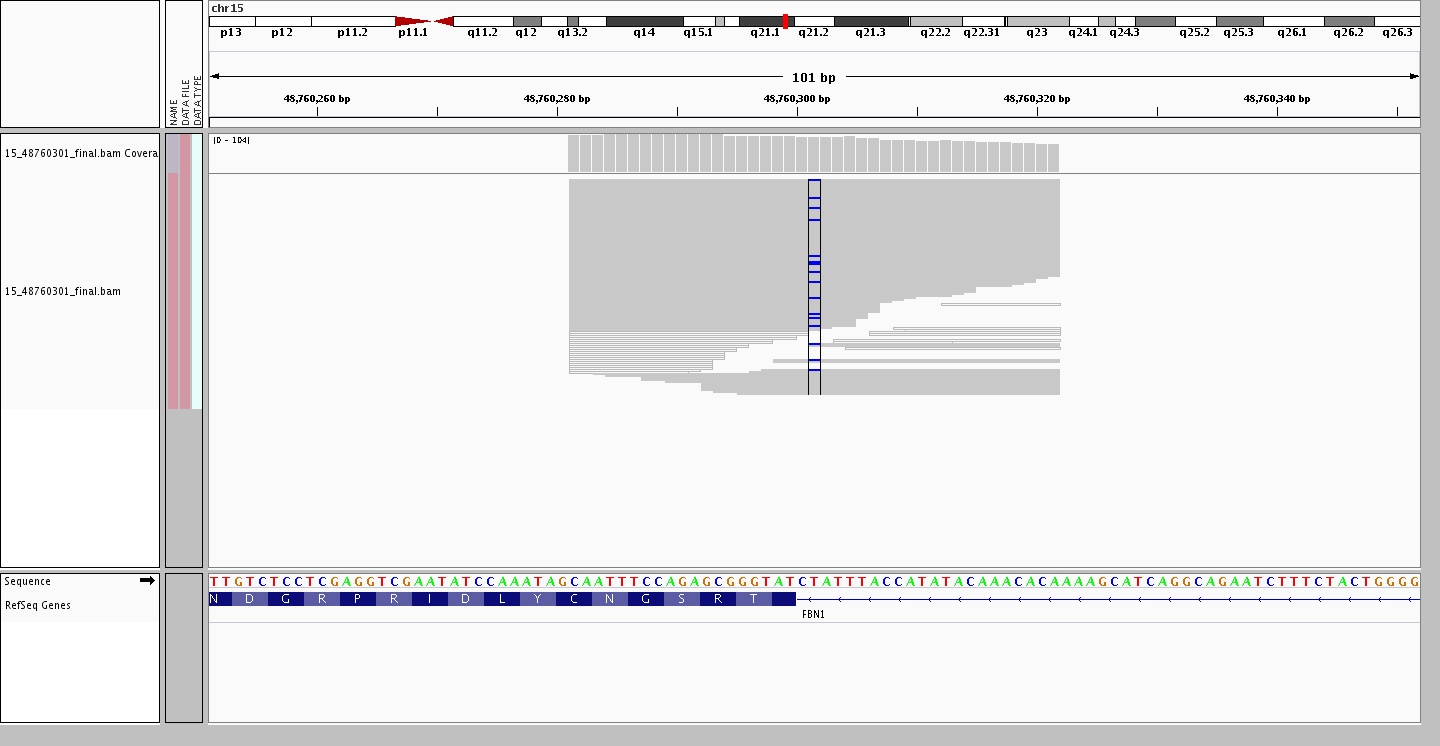
One of the upcoming features we are developing for the ExAC browser is the ability to view the sequencing reads from the reconstructed BAMs produced by the Genome Analysis Toolkit (GATK) Haplotype Caller using the –bamOutput option. For the splice acceptor variant 15-48760301-T-C, this is particular useful to not only show the reads/bases supporting the SNP calls but also the reference sequence context and whether the acceptor site is the canonical (i.e. ends in [T/C]AG).
Note: FBN1 is on the reverse strand
MECP2 Example
Loss of Function and ClinVar variants
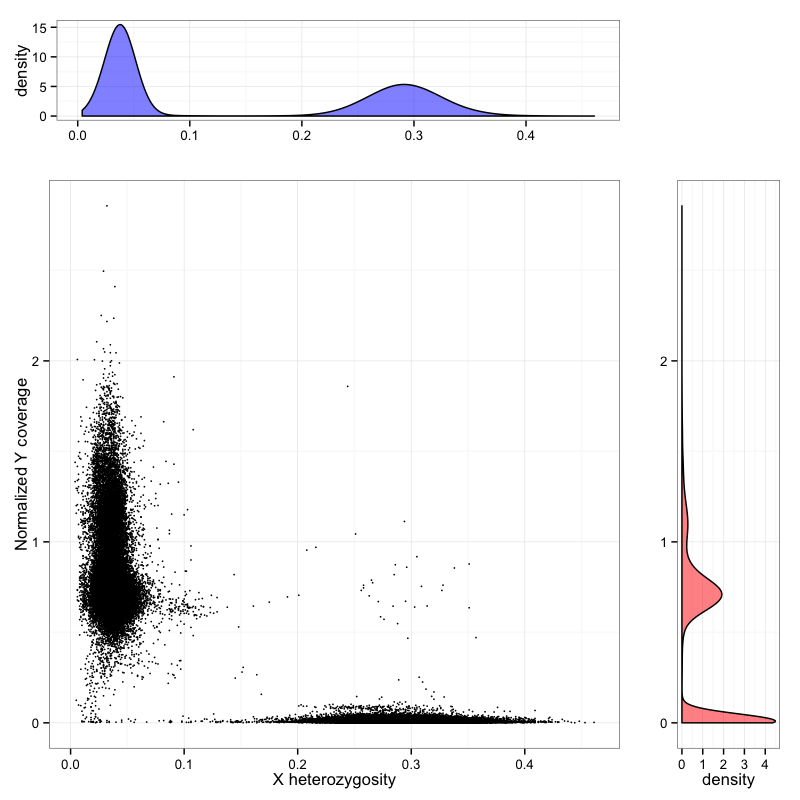
The LoF variants can be viewed by either following this link or searching MECP2 then clicking on the LoF button. In MECP2 there are 6 LoF variants. The stop gained variant X-153296689-G-A has an allele count of 68 with 20 homozygous individuals. Currently the ExAC data set is not sex aware and does not differentiate between hemizygous males and homozygous females. An upcoming feature is to calculate these numbers correctly for variants on the X chromosome. The sex of each individual in ExAC was determined by heterozygosity on the X chromosome and normalized chromosome Y coverage.
Differentiating males and females from exome sequencing data, using chrX heterozygosity (X axis) and coverage on the Y chromosome (Y axis). Males form a cluster on the left, females on the bottom right. A small number of unassigned individuals are also visible, some of whom are probable Klinefelter cases.
Now for the stop gained variant of interest, all 20 homozygous individuals are actually hemizygous males. Similar to the FBN1 example, the stop gained annotation only affects 1/3 transcripts while the other two (including the canonical) have a missense (p.Thr197Met, p.Thr209Met) annotation. According to ClinVar this variant is a missense variant and classified as benign. The LoF variants X-153296104-TCAGG-T and X-153296112-AGGTGGGG-A with homozygous individuals are also due to hemizygous males.
The variant X-153295997-C-T is an example of a pathogenic ClinVar variant in MECP2 that claims to be associated with neonatal severe encephalopathy in males. The 4 homozygous individuals in ExAC are actually 4 hemizygous males. It was soon later argued to be a rare variant rather than pathogenic but still remains classed as pathogenic in ClinVar!
Finally for a pathogenic variant in ClinVar not found on the ExAC browser with genomic coordinates X-153296806. The coverage data also provided for download shows that this site has adequate coverage for variants to be detected.
tabix -h Panel.chrX.coverage.txt.gz X:153296806-153296806
Fraction of samples at various coverage.
| Chr | Pos | Mean | Median | 1x | 5x | 10x | 20x | 30x | 50x | >=100x |
|---|---|---|---|---|---|---|---|---|---|---|
| X | 153296806 | 72.81 | 72.00 | 1.0000 | 1.0000 | 0.9995 | 0.9918 | 0.9670 | 0.8025 | 0.3005 |
Looking more deeply at the insertion/deletions (indels) that result in frameshifts
Another advantage of having the ability to view sequencing reads is that users can now look at the reliability of the more difficult indels and other complex variant calls. The Exome Variant Server (EVS) is a fantastic resource for the research community but did not have features for researchers to scrutinize indel variant calls. This was particularly concerning for researchers when publishing on novel disease gene. In the case of a recently published paper on LMOD3, for instance, the presence of homozygous frameshift indels in EVS greatly concerned our collaborators; it was only through careful scrutiny of the raw data for these variants that we were able to reassure them that these were genotyping errors.
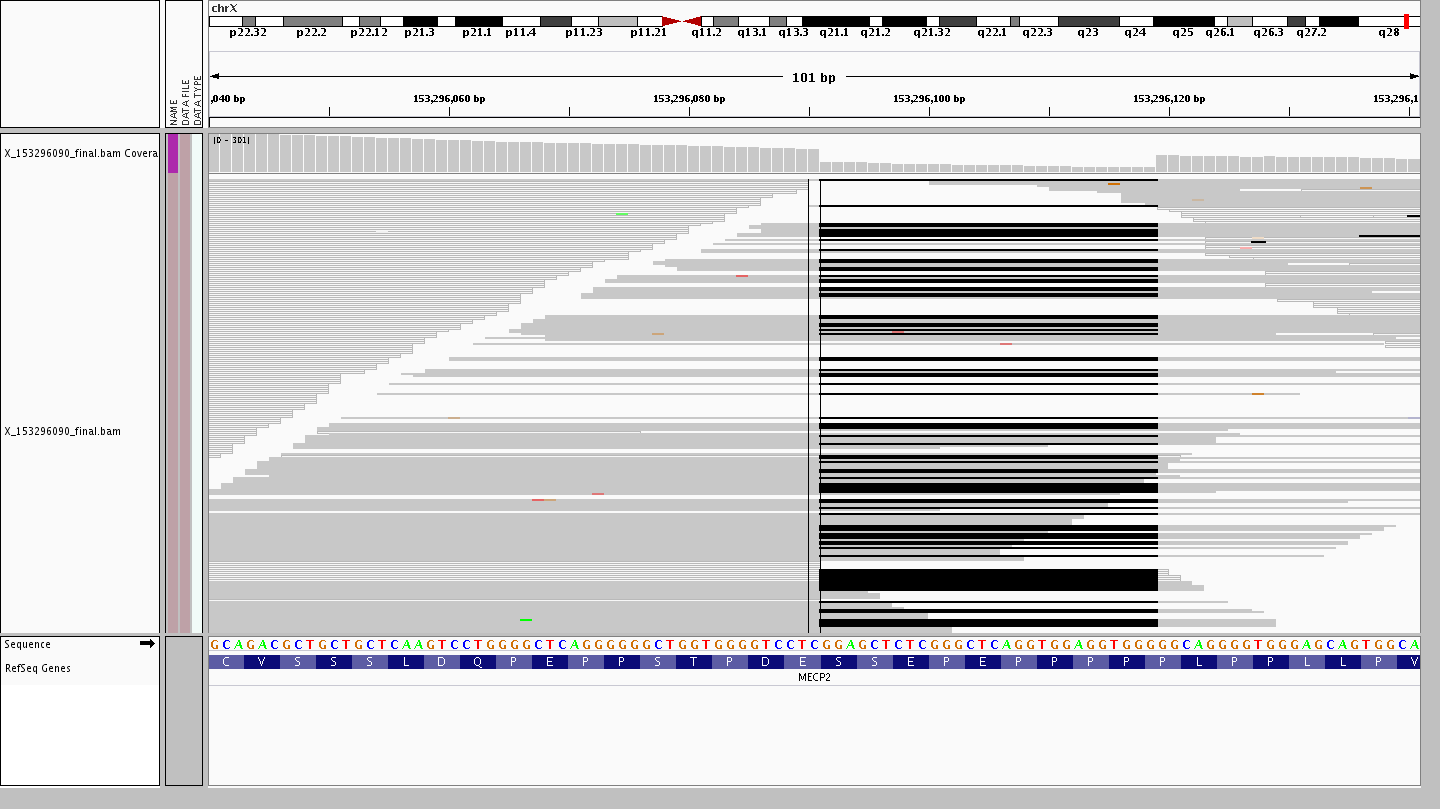
Firstly, let’s take a look at the 28 bp deletion X-153296090-CGGAGCTCTCGGGCTCAGGTGGAGGTGGG-C in MECP2 resulting in a frameshift variant.
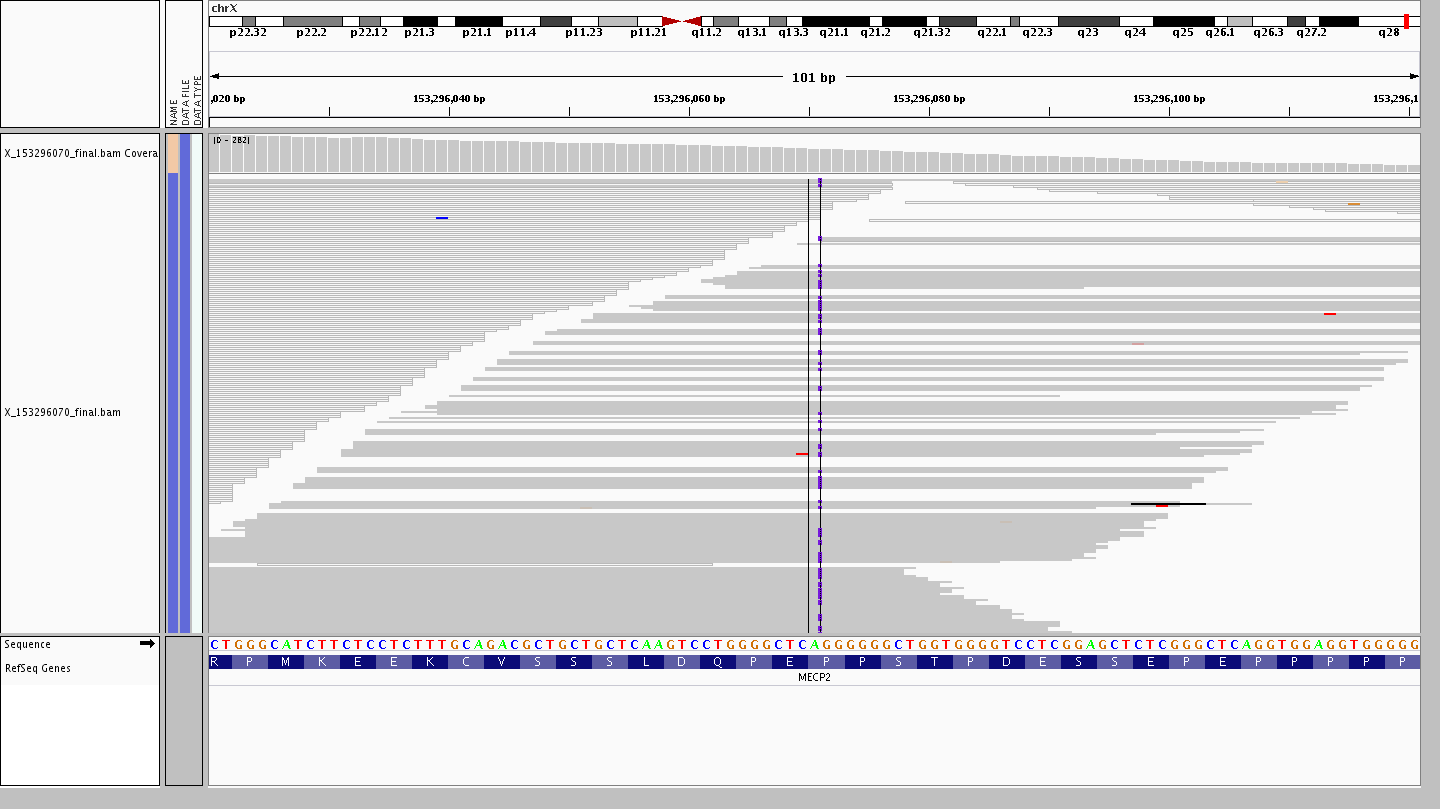
Now let’s see the reads for a 1 bp insertion X-153296070-A-AG from a heterozygous female.
Both of these frameshift mutations appear real and may cause intellectual disability, so why do they exist in a data set of individuals without severe diseases? I propose three possible reasons:
- There is an obvious drop in coverage where all the LoFs in MECP2 have accumulated. This may indicate a region difficult to capture or sequence and perhaps also challenging to detect variants.
- The shorter protein coding transcript ENST00000407218 avoids all but 1 LoF mutation (X-153296689-G-A) and may rescue some function lost in the larger isoforms.
- Lastly, the LoFs are towards the end of the gene and may result in a much milder phenotype.
Investigating which of these possibilities may be contributing will require further detailed analysis. We welcome comments from MECP2 researchers regarding the LoF mutations in ExAC.
Tri and Quad allelic SNPs
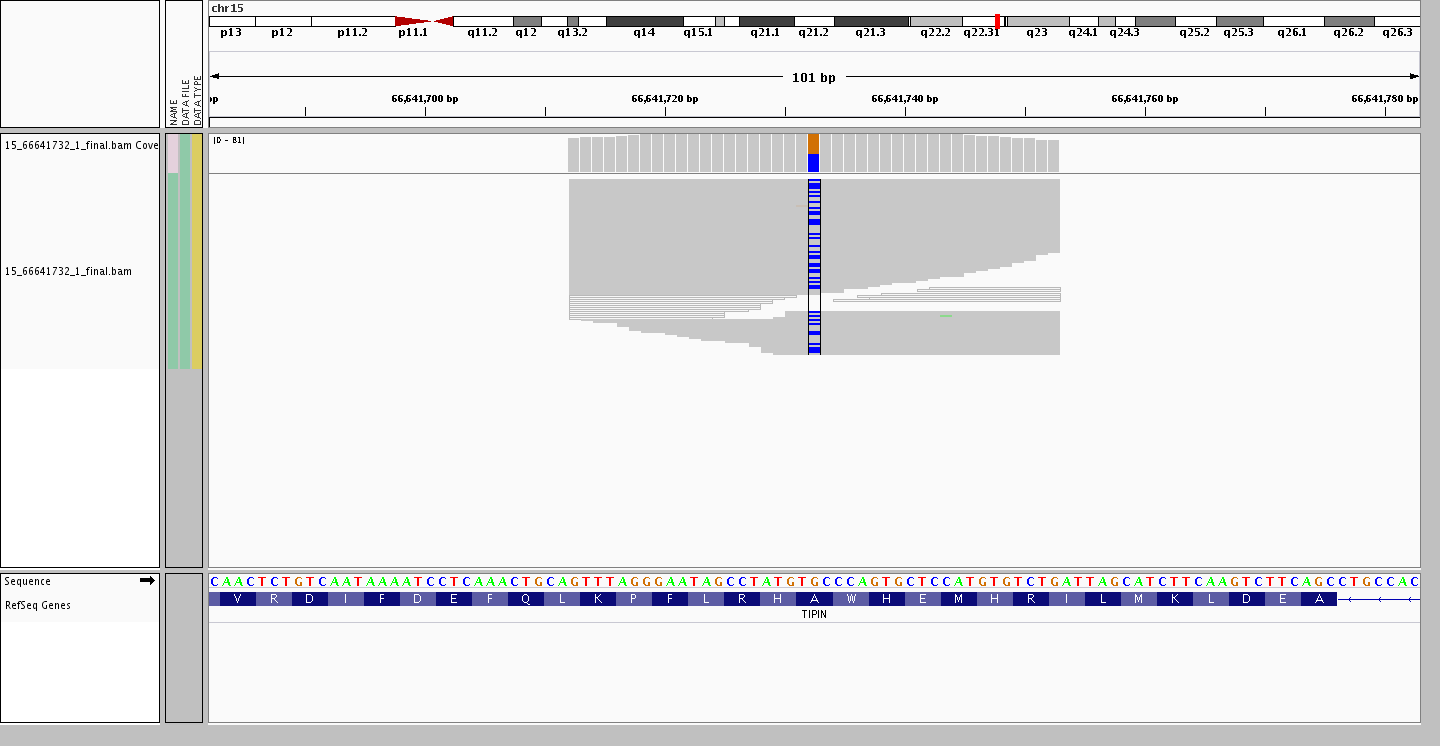
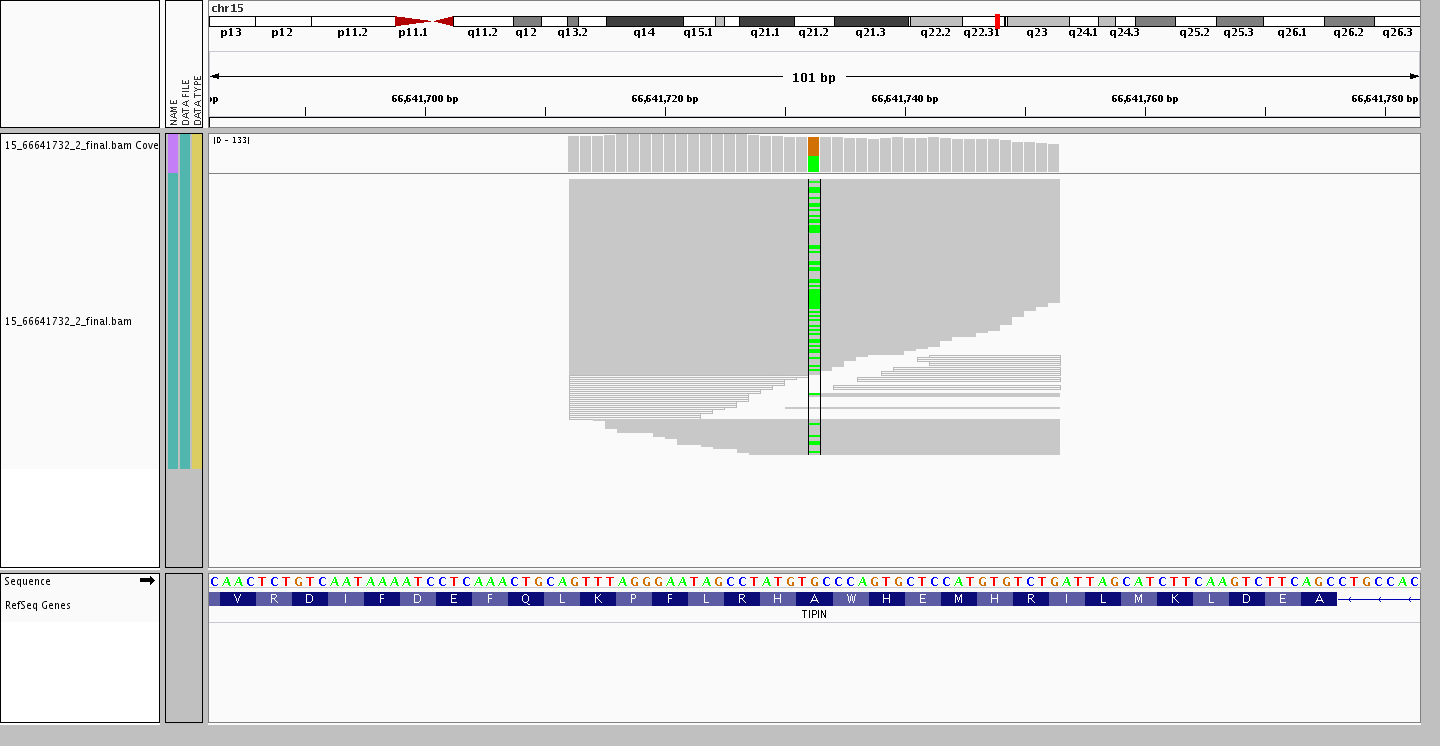
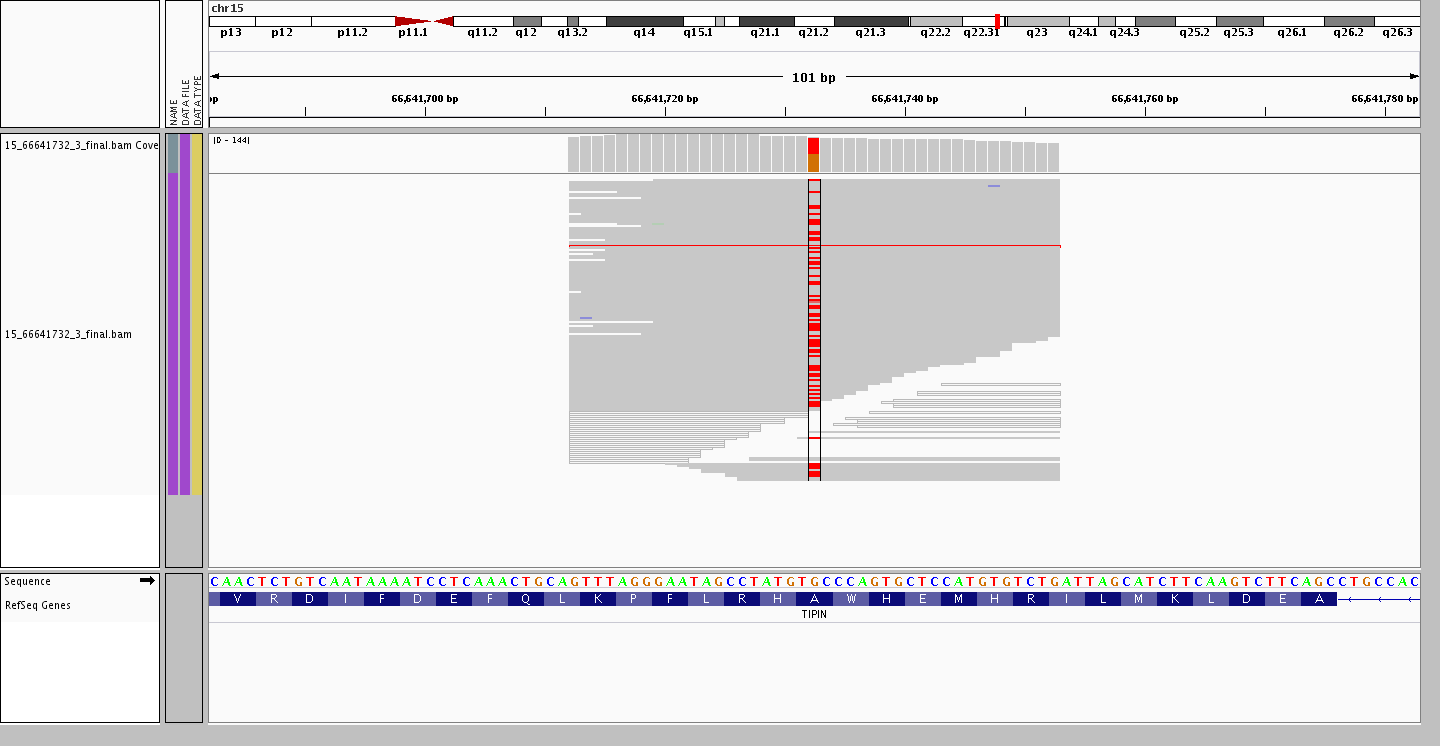
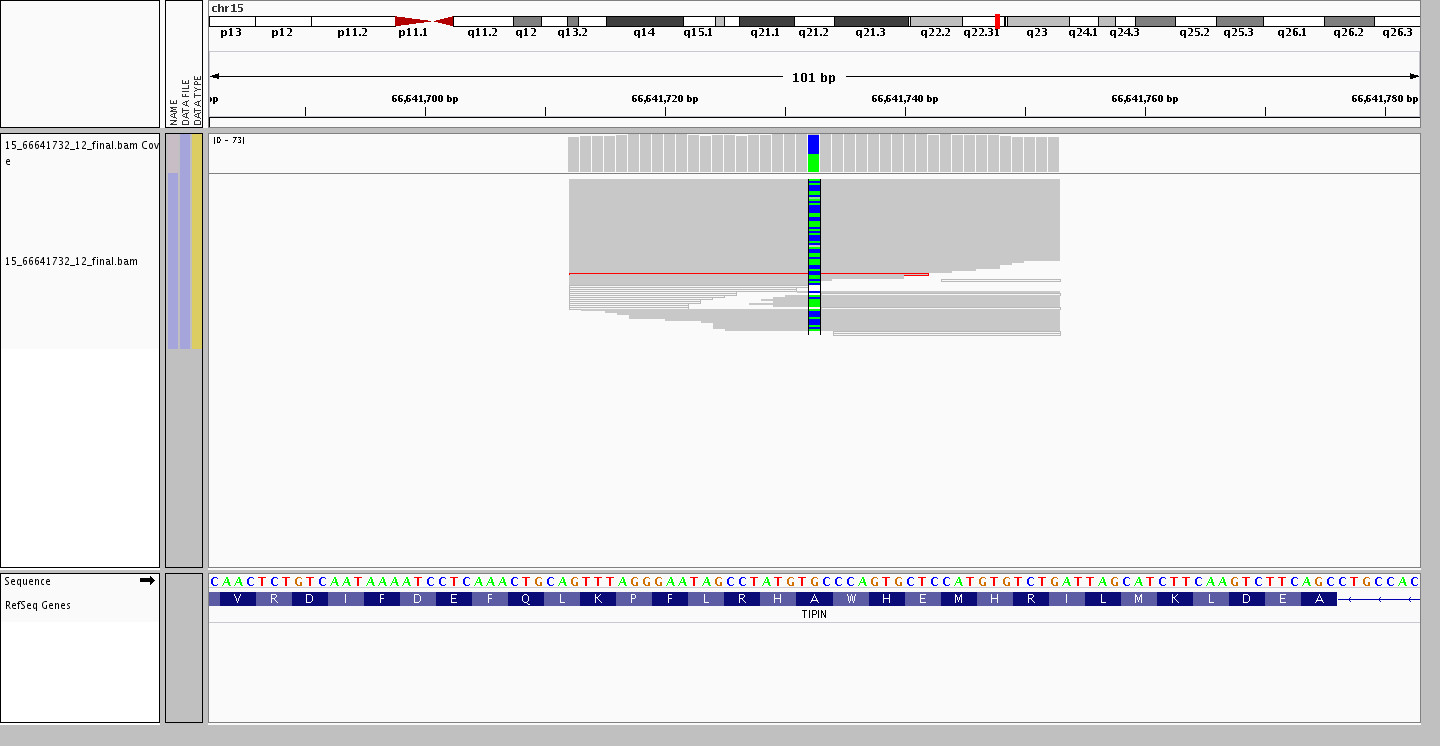
Ending on an interesting point resulting from larger and larger data sets. The assumption that common variants remain bi-allelic is no longer valid, as with each new individual added there is a possibility of finding a new allele at a site where a bi-allelic variant is present. For example, the variant site rs2063690 is now a quad-allelic SNP – in other words, every possible base is present at this site in at least one individual in our data set! Furthermore, the figures below shows three individuals who are heterozygous for the reference and each of the alternate alleles, while the last individual is heterozygous for two alternate alleles.
Heterozygous G/C (ref/alt)
Heterozygous G/A (ref/alt)
Heterozygous G/T (ref/alt)
Heterozygous C/A (alt/alt)
There is increasing urgency for the development of tools that deal appropriately with these mult-iallelic sites – approximately ~7% of ExAC sites are now multi-allelic, and that fraction will grow as our sample size increases. That high rate of multiallelism shouldn’t be surprising, by the way – the ExAC data set now (staggeringly) contains one variant every six bases on average, so it’s not a shock to see many cases where variant locations overlap.
Final thoughts
We’ve been gratified to see the rapid and positive response of the community to the ExAC data set. We still have plenty of work to do, though – and we’d love to get your feedback. If you have issues with the data set or the website, please drop us an email. For website bugs or feature requests you can also lodge a Github issue.
Many thanks to Daniel MacArthur for comments/feedback, writing introduction and final thoughts!

what is considered a rare variant in ExAC?
LikeLike
Hi Monkol,
I wonder if you could tell me how can i download variants of gene/s identified in a specific population with allele frequencies.
LikeLike
Hi Monkol, what does the HEMI field in the database mean? I am looking at recessive infant/childhood onset diseases and was wondering if i can reject “known pathogenic variants” in sex-linked diseases if there are hemizygous individuals reported in ExAC for those particular variants?
Thank you for the clarification.
LikeLike
The ftp site is down, and the data are currently unavailable.
LikeLike
Tried to send email to exac.data@gmail.com and got the following message back
exac.data@gmail.com: 550 5.2.1 The email account that you tried to reach is disabled. l65si22905737iod.75 – gsmtp
LikeLike
Our bad! Have updated the email link to the one you should use to get in contact. Sorry for the inconvenience.
LikeLike
It would be nice if ExAC website also included instructions on how to use this so that less proficient people like most neurologists ( like myself) can make use of it. Thank you for the great work.
LikeLike
Thanks to the sequencing technology so that we can have a clear understanding on human beings. Thanks to your analysis and contribution to this post so that I can learn things about exome data set!
LikeLike